L’étude Xeroderma pigmentosum teste des oligonucléotides synthétiques hypoallergéniques en tant qu’agent thérapeutique
Des chercheurs en génétique de l’Université de Nagoya, au Japon, ont étudié la base génétique d’une affection cutanée rare affectant les enfants et extrêmement courante au Japon.
Dans le document de recherche, « Les mutations fondatrices introniques profondes identifiées dans le gène ERCC4/XPF sont des cibles thérapeutiques potentielles pour une forme à haute fréquence de xeroderma pigmentosum » publié dans PNASÀ l’aide d’oligonucléotides synthétiques anti-allergiques, l’équipe trouve une cible thérapeutique potentielle pour la maladie.
Xeroderma pigmentosum (XP) est une maladie génétique rare qui peut entraîner une sensibilité accrue au soleil et un risque accru de tumeurs cutanées en raison d’une déficience du système de réparation de l’ADN responsable du traitement de la lumière du soleil.
Les patients XP présentent généralement des problèmes cutanés graves, notamment une photosensibilité, une peau sèche, des anomalies de la pigmentation cutanée et un risque accru de cancer de la peau. Certains cas peuvent également présenter des symptômes neurologiques.
Les manifestations cliniques de XP varient en fonction des gènes affectés et des types de mutations. Alors que chaque forme de XP montre une certaine maladie, les patients XP-F peuvent avoir la plupart ou toutes les manifestations de la maladie simultanément.
La pénétration globale de XP est rare à environ 3 par million. Au Japon, les taux sont beaucoup plus élevés, 1 sur 22 000. Parmi les cas de XP, les incidents XP-F représentent environ 1 % dans le monde et 4 % au Japon, ce qui rend le XP-F 66 fois plus courant au Japon que la moyenne mondiale .
En partie parce que XP est une maladie si rare, elle est toujours à l’étude et les options de traitement sont limitées. Il est nécessaire d’améliorer la compréhension, le pronostic et les cibles thérapeutiques potentielles pour XP, en particulier pour les variants rares tels que XP-F.
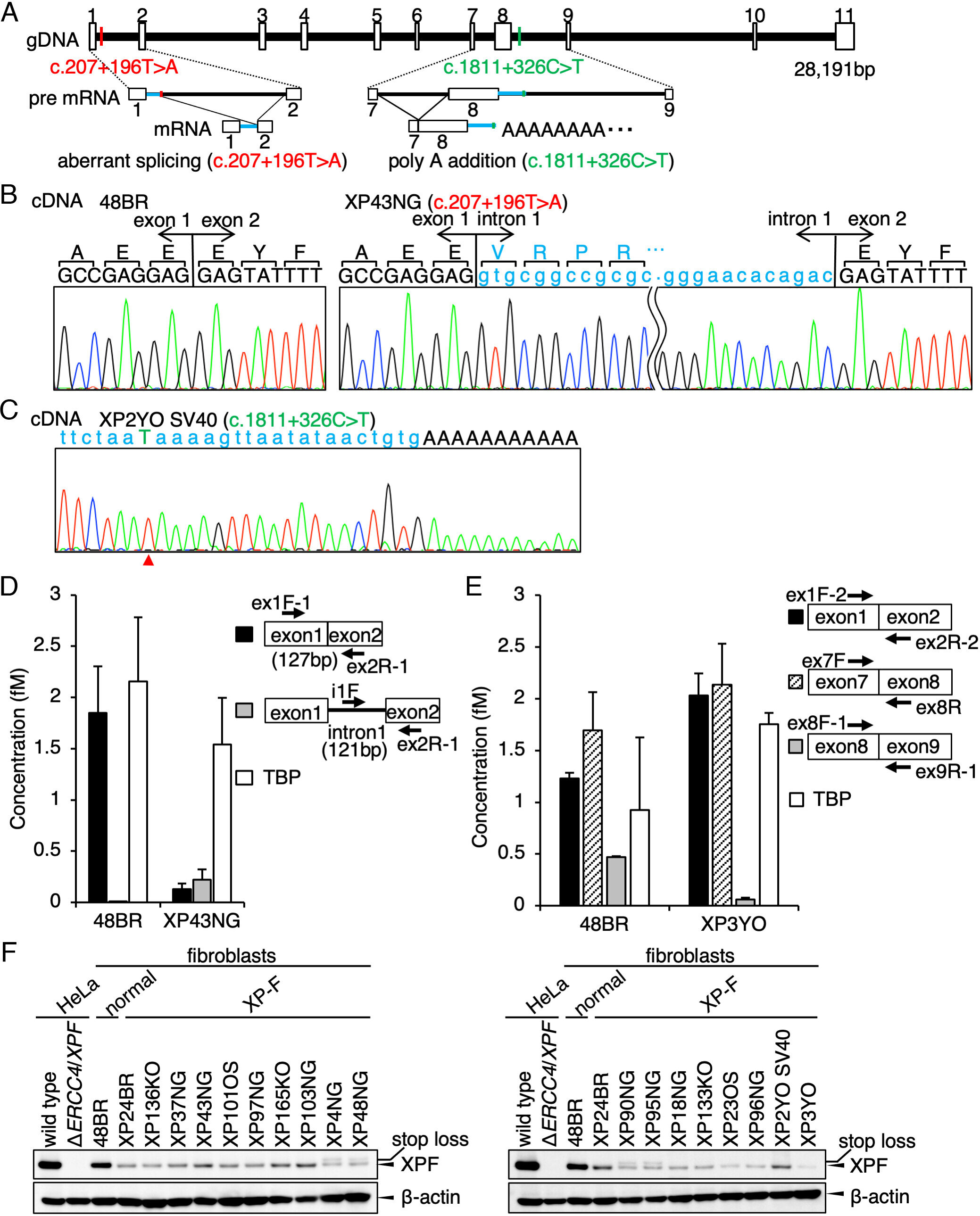
L’étude a été menée dans une cohorte japonaise XP (taille de la cohorte définie comme « la plus grande » uniquement) et a identifié 17 cas de XP-F, tous hébergeant l’une des deux variantes du gène ERCC4/XPF. La première variante est une mutation fondatrice japonaise qui représente environ 10 % de tous les cas de XP japonais et provoque un épissage pré-ARNm incorrect. La deuxième mutation induit un substitut de polyadénylation.
Les deux mutations entraînent une diminution de l’expression du gène ERCC4/XPF, ce qui entraîne une diminution significative de l’expression de la protéine XPF et un déficit de réparation de l’ADN dans les cellules des patients.
L’équipe a tenté de corriger des événements d’épissage anormaux dans des échantillons de cellules de patients avec des oligonucléotides antisens synthétiques (ASO). Les ASO fonctionnent en se liant à des séquences d’ARNm spécifiques. Ils peuvent dégrader ou modifier l’épissage, empêcher la traduction ou, dans ce cas, interférer avec des événements alternatifs de traitement de l’ARNm, qui peuvent empêcher ou réguler la production de protéines en aval.
Après traitement avec des ASO, l’expression de la protéine XPF a été récupérée au niveau normal, indiquant que les ASO ont efficacement restauré l’expression de l’ARNm. L’étude démontre que les oligonucléotides antisens spécifiquement conçus pour ces mutations peuvent restaurer l’expression de la protéine XPF et la capacité de réparation de l’ADN dans les cellules des patients XP-F.
Alors que les ASO sont actuellement développés et testés pour une variété de maladies à base génétique, l’application dans l’étude actuelle montre comment les études génétiques peuvent ouvrir la voie à la recherche de cibles thérapeutiques même pour les maladies les plus rares.
Plus d’information:
Chikako Senju et al, Les mutations fondatrices introniques profondes identifiées dans le gène ERCC4/XPF sont des cibles thérapeutiques potentielles pour une forme à haute fréquence de xeroderma pigmentosum, Actes de l’Académie nationale des sciences (2023). DOI : 10.1073/pnas.2217423120
Informations sur la revue :
Actes de l’Académie nationale des sciences
© 2023 Réseau Science X