Une méthode de modélisation de la décomposition d’un électrolyte polymère lors du processus de nucléation du lithium dans les batteries au lithium métal

Figure 1. Organigramme pour la simulation d’un système d’anode PEO-lithium. Les calculs totaux ont été effectués en quatre étapes. Tout d’abord, une simulation AIMD 5 ps de SPE sur la surface de l’anode Li a été réalisée. Ensuite, 10 atomes de Li ont été ajoutés et pré-calibrés pendant 500 fs pour trouver des positions initiales plausibles pour ceux-ci tandis que le reste du système était fixe, et des simulations AIMD ont été effectuées 3 ps plus tard. Cela imite Li+ réduction pendant le placage au lithium. L’addition et la simulation d’atomes de Li ont été répétées deux fois de plus, pour un total de 14 calculs AIMD, ajoutant un total de 30 atomes de Li. La température a été fixée à 600 K pendant la période de pré-équilibre pour accélérer la relaxation des atomes de lithium ajoutés. Après pré-équilibrage de chaque phase, le polymère et les trois couches supérieures de la surface de l’anode Li ont été relâchées, la simulation AIMD du système d’anode PEO-Li a pu continuer. Les charges électroniques ont été calculées à l’aide de l’analyse de charge Bader29Et3031. Dix configurations dans les 500 dernières fs proches du minimum d’énergie ont été sélectionnées pour obtenir la charge atomique à chaque étape. Les atomes de différents environnements chimiques ont été distingués et les charges atomiques ont été calculées. La distribution de la charge atomique pour chaque environnement chimique a été donnée par une fonction gaussienne.
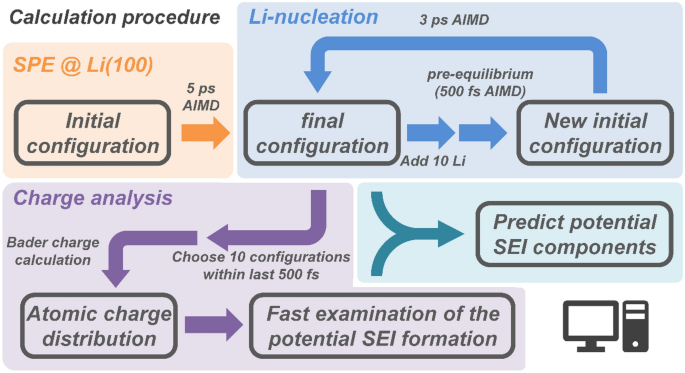
Effectuez le calcul général pour prédire les composants SEI potentiels.
La figure S1 affiche le profil énergétique de simulation AIMD du PEO pur. Les composants PEO n’ont subi aucune dégradation au cours de la simulation AIMD 10 ps, et le système a atteint l’équilibre thermique apparent en 1 ps. Comprendre le mécanisme de dégradation du PEO observé lors de précédentes mesures XPS in situ26où l’énergie de liaison est calibrée en utilisant le pic de 286,7 eV à C 1s Spectre PEO À titre de référence, nous avons effectué des simulations AIMD en quatre phases, comme illustré à la Fig. 1. Le diagramme d’énergie de PEO sur un système Li-anode pendant le temps de simulation est illustré à la Fig. 2a. La première phase (régions jaunes) représente le PEO sur la surface de l’anode Li(100) ; L’énergie atteint immédiatement l’équilibre thermique apparent, ce qui signifie que la décomposition du PEO ne s’est pas produite à ces échelles de temps. Après avoir simulé 5 ps, nous avons simulé le processus de nucléation du Li en trois phases supplémentaires (régions bleue, verte et rouge). Dix atomes de lithium supplémentaires ont été ajoutés à la configuration finale de l’étape précédente. Une procédure de pré-équilibrage supplémentaire a été effectuée et la fluctuation d’énergie est illustrée à la Fig. S2. Pour chaque étape de la nucléation de Li, le système atteint un équilibre thermique apparent dans la simulation AIMD 3 ps, et l’énergie dans chaque étape diminue, ce qui signifie que la décomposition du PEO s’est produite. Les simulations indiquent que la réduction et le dépôt de Li dégradent le PEO beaucoup plus que la simple exposition de l’électrolyte à base de PEO à la surface du Li-métal.
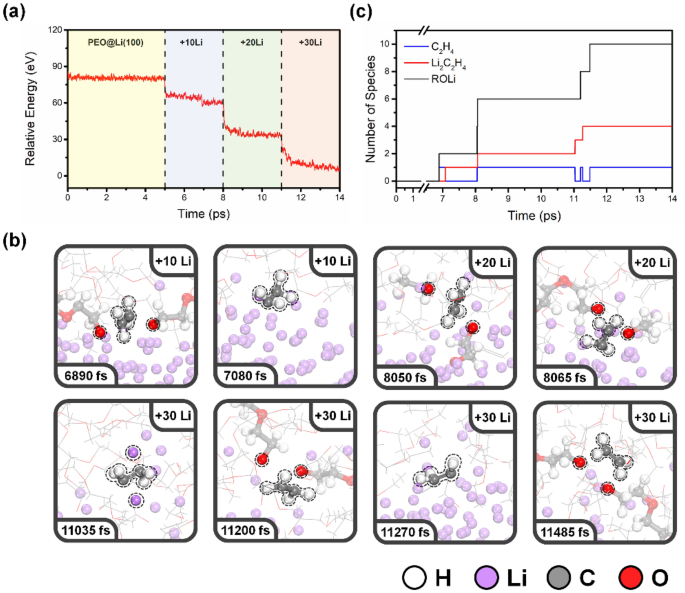
(une) Diagramme d’énergie pendant le temps de simulation du PEO sur la surface de l’anode Li (100) pendant la nucléation de Li. (B) Captures d’écran de PEO sur la surface de l’anode Li(100) à partir de simulations AIMD. (c) le nombre de produits de décomposition obtenus lors des réactions de décomposition du PEO sur la surface de l’anode Li.
La figure 2b affiche une série d’instantanés à différents moments de simulation extraits de la simulation AIMD. La première réaction de décomposition du PEO est observée à 6890 fs, dans la première étape de Li-nucléation. La première chaîne de PEO a été hydrolysée, formant deux alcoxydes de lithium, l’un des principaux composants SEI observés pour la dégradation du PEO à la surface du Li25,26. De plus, une seule molécule d’éthylène peut être formée par deux fentes de liaison C – O à 7080 fs. La molécule d’éthylène hautement réactive formée a interagi avec la surface métallique du lithium et a généré des complexes lithium-éthylène (Li2c2h4)32. Une séquence de réactions de décomposition du PEO a ensuite été observée à 8050, 8065 et 11200 fs, et les molécules d’éthylène résultantes ont ensuite formé Li2c2h4 à 11 035, 8 065 et 11 270 fs, respectivement. La dernière décomposition du PEO s’est produite à 11485 fs, où la molécule d’éthylène est restée en vue du système s2 Hybridation jusqu’à la fin de la simulation AIMD. Toutes les interactions interfaciales et les heures approximatives des événements sont résumées dans le tableau S1. Les produits de décomposition résultants comprennent des oxydes de lithium (Rolli), des molécules d’éthylène (C2h4) et Li2c2h4, et la formation d’espèces de dégradation à l’interface anode PEO-Li. Le nombre de ces espèces est indiqué sur la figure 2c, dans laquelle se trouvent les alcoxydes de lithium et de Li2c2h4 sont des composantes majeures du SEI. Les molécules d’éthylène sont généralement formées comme intermédiaires, mais sont présentes en présence de Li0 directement à moi2c2h4. Il est important de noter que les simulations AIMD explorent qualitativement les produits de décomposition potentiels et les mécanismes de réaction potentiels et fournissent le moment relatif plutôt qu’absolu des interactions.
La distribution de la charge atomique des atomes d’oxygène et de carbone dans le système d’anode PEO et PEO-Li à différents stades de la nucléation de Li est présentée sur les figures 3a et c, respectivement. Pour les atomes d’oxygène, un seul environnement chimique existe dans le PEO pur, c’est-à-dire l’oxygène de l’éther (OC, ~−1,02 | e |). Après avoir placé du PEO sur la surface de l’anode Li, la distribution reste presque la même, mais le centre du pic est légèrement décalé vers des valeurs plus négatives (~- 1,05 |e|), ce qui est dû aux atomes d’oxygène de la chaîne PEO s’adsorbant à la surface de l’anode de lithium, cela fait que les charges atomiques des atomes d’oxygène deviennent plus négatives. Cette pente plus faible est également observée en O 1s Spectre XPS de PEO, dans lequel un déplacement de la position du pic a été observé de 532,7 à 532,6 eV lorsque du lithium a été déposé à la surface du PEO. Un pic supplémentaire est observé dans la plage -1,3 à -1,4 | e | Cela indique la formation d’une nouvelle espèce, correspondant à l’alcoolate de lithium (ROLi), tandis que les atomes d’oxygène gagnent en densité électronique à partir du lithium métallique. Dans le même temps, un nouveau pic d’O expérimental 1s Le spectre XPS à 530,4 eV étudie la formation d’espèces d’alcoxyde de lithium. L’intensité du pic d’oxyde de lithium augmente progressivement après les deux processus de nucléation du lithium suivants dans la simulation. Deux pics sont présentés dans le système PEO pur d’atomes de carbone. Le pic principal est ~ + 0,4 | e | correspond aux atomes de carbone du squelette PEO (C–O), qui est également observé pour C1s Spectres XPS à 286,7 eV. Le petit pic est à ~ – 0,1 | e | Il correspond à la fin de la chaîne PEO (–CH3), qui ont le même environnement chimique que les hydrocarbures (C – C / C – H), et empirique C 1s Signal du spectre XPS à 285,1 eV qui correspond à une contamination de surface par des hydrocarbures lors de la préparation des échantillons. Dans la simulation, la concentration de –CH3 Les groupes sont beaucoup plus élevés que leurs homologues expérimentaux, pour des raisons pratiques. Au cours des processus de nucléation du Li, deux nouveaux pics à ~−0,1| ont été générés e | et ~ – 0,7 | e | ; Les environnements chimiques correspondants sont C2h4 Gardien2c2h4 espèces, respectivement. Considérant la position du pic analogue à (–CH3) et C2h4, ceux-ci sont susceptibles de fusionner et d’apparaître sous la forme d’un pic unique mais avec un petit élargissement, comme indiqué par la croissance du pic CC / CH sur la figure 3d. Littérature précédente21,25 Montre que la présence d’eau peut affecter la dissolution de l’électrolyte. Cependant, l’absence de pic LiOH sur la figure 3b montre que la teneur en eau est négligeable, ce qui peut être attribué à l’état de vide poussé dans la mesure XPS (~ 10– dix mbar) et une expérience Li-Deposition (~10–8 mbar). Comme mentionné ci-dessus, la distribution de charge atomique est un descripteur efficace pour comprendre les pics dans le spectre XPS.
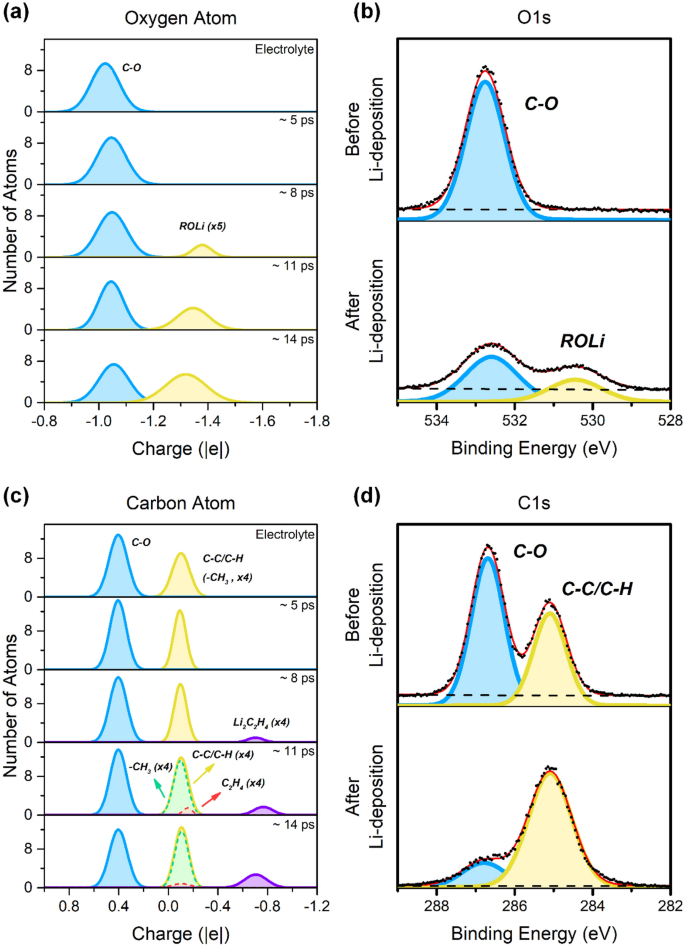
La distribution de la charge atomique de (une) oxygène et (c) les atomes de carbone dans le PEO pur et sur la surface de l’anode Li(100) sont à différents stades de nucléation du Li. référence (B) Q1s Et (DocteurC 1s Spectres XPS de PEO pur avant et après la nucléation de Li, en utilisant des données précédemment publiées dans la réf.26.
Pour étudier en détail le rôle de Li supplémentaire, nous avons placé l’oligomère PEO dans une couche sous vide et l’avons adsorbé sur une surface Li(100). Nous avons également adsorbé un composite PEO-Li sur une surface Li(100), qui comprend 5 couches de Li avec une supercellule 5 × 5. La figure S3 montre les structures optimisées du PEO avant et après adsorption sur la surface Li(100) et du composite PEO-Li adsorbé sur la surface Li(100). La densité d’états prédite correspondante (PDOS) pour le PEO dans chaque système est illustrée à la figure S4. Après adsorption du PEO sur la surface Li, sa bande de conduction, qui est au-dessus du niveau de Fermi, est fortement hybridée avec la surface Li et élargie au niveau de Fermi, ce qui explique le transfert d’électrons de la surface Li vers le PEO. De plus, la bande de conduction du complexe PEO-Li s’est déplacée davantage lorsqu’il s’est adsorbé sur la surface Li(100). Le déplacement vers le bas de la bande de conduction du PEO après complexation avec Li indique que du Li supplémentaire facilite le transfert d’électrons de la surface du Li vers le PEO. Ceci est vérifié par les frais calculés pour PEO. Initialement, le PEO porte une charge nulle dans une couche de vide. Après adsorption sur la surface Li, il gagne des électrons de la surface Li, résultant en une charge Bader de 0,56 | e | pour PEO. De plus, lorsqu’il est complexé avec Li, il montre une plus forte tendance à recevoir des électrons de la surface de Li, ce qui entraîne une charge Bader de -0,74 | e | pour PEO. Étant donné que le complexe PEO-Li a une bande de faible conductivité, le complexe présente une réactivité plus élevée que l’oligomère PEO pur et subit une hydrolyse réductrice lors des simulations AIMD.
Cependant, la nature des atomes de lithium en excès reste incertaine. Par conséquent, nous avons remplacé l’atome Li supplémentaire par un ion Li supplémentaire, formant PEO-Li+ Un ion complexe adsorbé sur la surface Li(100), comme illustré à la Fig. S5a. Nous avons trouvé que les structures de PEO-Li et PEO-Li+ Ils étaient presque identiques lorsqu’ils étaient adsorbés sur une surface de Li(100). Géométrie de PEO-Li @ Li(100) et PEO-Li+ Les systèmes @Li (100) sont représentés respectivement par les modèles de balle et de bâton noir et rouge sur la figure S5b. De plus, le coût Bader calculé pour PEO est de – 0,74 | e | et – 0,67 | e | en PEO-Li@Li(100) et PEO-Li+ @ Système Li(100) d’affilée. Li supplémentaire dans les deux systèmes montre une charge Bader de +0,88 | e | , indiquant son caractère Li-ion. Cela indique que Li supplémentaire transfère immédiatement des électrons à la chaîne PEO, formant des ions Li et des électrons en excès. La complexation des ions Li avec le PEO abaisse l’énergie de la bande de conduction du PEO, tandis que les électrons en excès remplissent la bande de conduction inférieure du PEO. Bien que PEO-Li+ Le système @Li(100) porte une charge positive et le PEO reçoit toujours des électrons de la surface Li où l’anode Li-métal agit comme source d’électrons. Par conséquent, l’effet de l’électron supplémentaire est négligeable. Le rôle des atomes de Li coordonnés dans les électrolytes liquides a été largement étudié dans des études informatiques33Et3435, et ces résultats sont également applicables aux électrolytes polymères solides. Compte tenu de cela, l’approche de simulation utilisée dans cette étude peut également être appliquée au modèle de décomposition interfaciale dans les batteries Li-métal à l’état solide, où les ions lithium et les électrons migrent vers la surface de l’anode via l’électrolyte et les circuits externes, respectivement. Les processus du complexe PEO-Li et le transfert d’électrons en excès vers le PEO sont essentiels à la fois pour le dépôt de Li et la charge de la batterie Li-métal.



")
